About Pompe disease
Pompe disease pathophysiology and natural history
Pompe disease, also referred to as acid maltase deficiency (AMD) or glycogen storage disease type II (GSDII), is a rare autosomal recessive lysosomal storage disorder (LSD) caused by mutations in the GAA gene coding for the enzyme acid alpha-glucosidase (GAA).1,2
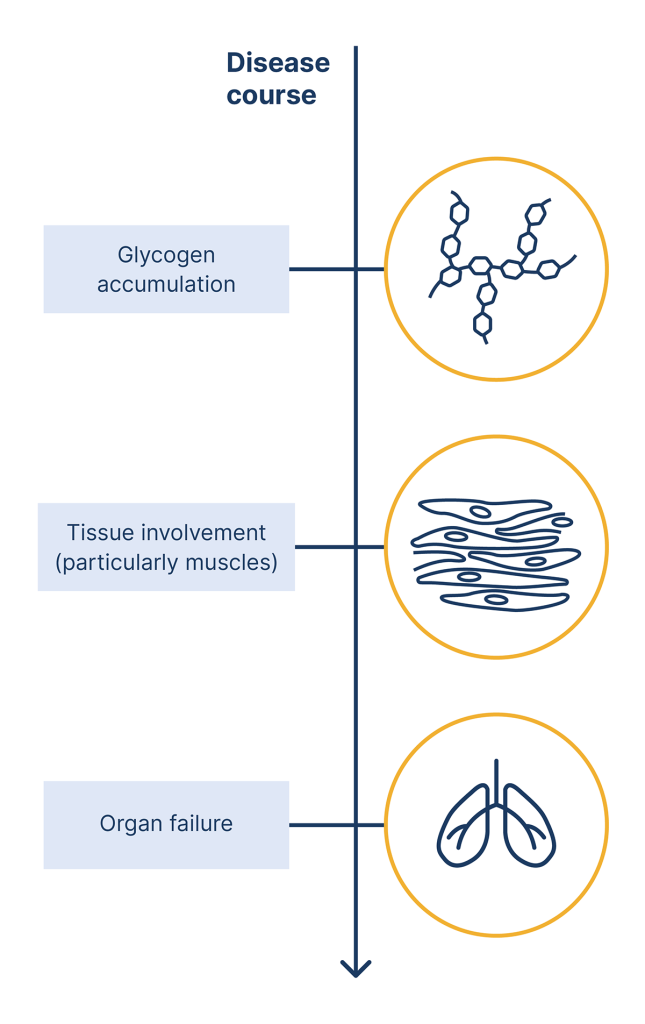
The figure below summarises the underlying mechanism of Pompe disease.
Pompe disease is caused by mutations or variations in the GAA gene
These mutations can lead to absence or defiency of an enzyme called acid alpha-glucosidase (GAA)
When GAA is absent or deficient, its substrates glycogen, accumulates in the lysosomes of cells
This leads to cell damage within affected tissues, and causes the various pathologies seen in Pompe disease
Underlying mechanism of Pompe disease.2,3
Pompe disease subtypes
A wide range of phenotypic variability exists in Pompe disease. This had led to the creation of two broad subtypes – ‘infantile onset’ and ‘late onset’ – based on the age of onset and degree of organ involvement.1,3
Click on each heading to learn more about the main subtypes of Pompe disease.
- Presents in the first year of life
- Little or no GAA activity (<1% of normal)
- Key signs and symptoms:
- Muscle hypotonia
- Cardiomyopathy
- Respiratory insufficiency
- Most untreated patients die within one year
- Presentation can vary from the first year of life to the eighth decade
- Residual GAA activity (<30% of normal)
- Key signs and symptoms:
- Progressive muscle weakness
- Respiratory abnormalities
- Some patients not diagnosed for over 10 years after onset of clinical signs
Please note, these subtypes can be further subcategorised according to the degree of organ involvement.1
The remainder of this resource will focus on the late-onset subtype of Pompe disease unless otherwise stated.
Natural history
Disease severity typically correlates inversely with residual GAA activity in cells and tissues. Late-onset disease symptoms are related to progressive weakness of the axial, limb-girdle and respiratory muscles, particularly the diaphragm.5
The life expectancy of patients with late-onset Pompe disease varies from early childhood to late adulthood, depending on the rate of disease progression, extent of organ involvement and other comorbidities.1 Patients may gradually lose their ability to walk and require mobility assistance, often via a wheelchair. Approximately half of these patients will suffer respiratory involvement and 30–40% will require treatment with ventilation in the later stages of disease.7 Respiratory failure remains the most common cause of death in late-onset Pompe disease.7

If you suspect Pompe disease, referral to nominated ultra-specialist centre is essential so that patients can access full genetic testing and access to disease-specific therapies. Please refer to our ‘Find a specialist centre’ page to find out more.
NP-NN-GB-00011223
October 2024
- Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-288.
- NORD. Pompe Disease. 2024. Available at: https://rarediseases.org/rare-diseases/pompe-disease/. Accessed: October 2024.
- Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Curr Treat Options Neurol. 2022;24(11):573-588.
- Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics. 2018;15(4):928-942.
- Toscano A, Rodolico C, Musumeci O. Multisystem late onset Pompe disease (LOPD): an update on clinical aspects. Ann Transl Med. 2019;7(13):284.
- Reuser AJJ, van der Ploeg AT, Chien YH, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum Mutat. 2019;40(11):2146-2164.
- Shah NM, Sharma L, Ganeshamoorthy S, Kaltsakas G. Respiratory failure and sleep-disordered breathing in late-onset Pompe disease: a narrative review. J Thorac Dis. 2020;12(Suppl 2):S235-S247.

0.
About Pompe

2.
Prevalence of Pompe disease