Neurology
Pathophysiology of muscle damage
The information on this page is tailored for neurologists. For more detailed information on the cause, inheritance, diagnosis and management of Pompe disease, please refer to our ‘About Pompe disease’ page.
Pathology in late-onset Pompe disease arises from the abnormal accumulation of glycogen in the lysosomes of cells throughout various tissues in the body, particularly the skeletal muscle.1
This excessive glycogen storage triggers lysosomal rupture, autophagy and consequent muscle damage.1
Click on the headings to find out more.
Glycogen accumulation causes lysosomes to swell and rupture in the intermyofibrillar space of skeletal muscle. Glycogen and other enzymes leak into the cytoplasm of the myocytes, impairing their contractile function and leading to muscle damage and decline of muscle function.2,3
Normal lysosomal glycogen degradation begins with autophagy, a normal process by which the body consumes its own damaged tissue. Double-membrane vesicles called autophagosomes form and collect cellular contents, after which they fuse with lysosomes to begin the degradation process.4 This fusion is often defective in Pompe disease, resulting in the accumulation of autophagosomes and other debris that impact the contractility of affected skeletal muscle fibres.1,2,5
Microscopic examination of muscle biopsies reveals cellular swelling and glycogen granules in muscle cells as well as excess glycogen in capillary walls of muscles and the skin.6 In the example below, you can see glycogen accumulation disrupting the myofibrils in skeletal muscle of a late-onset Pompe patient.

Scale bar: 0.5 μm.
Katona I, Weis J and Hanisch F. 2014, licensed under CC BY 2.0.7
Other pathogenic mechanisms contribute to muscle damage in late-onset Pompe disease, including dysregulated calcium homeostasis, oxidative stress and mitochondrial abnormalities.2,5
Progressive muscle weakness in Pompe disease
Muscle weakness is usually greatest proximally and impacts the lower extremities more than the upper extremities.8 The abdominal muscles, paraspinal muscles (with exception of the neck extensors and neck flexors), hip flexors, hip extensors, hip adductors and hip abductors are the most severely affected in late-onset Pompe disease, as shown in the diagram below.9
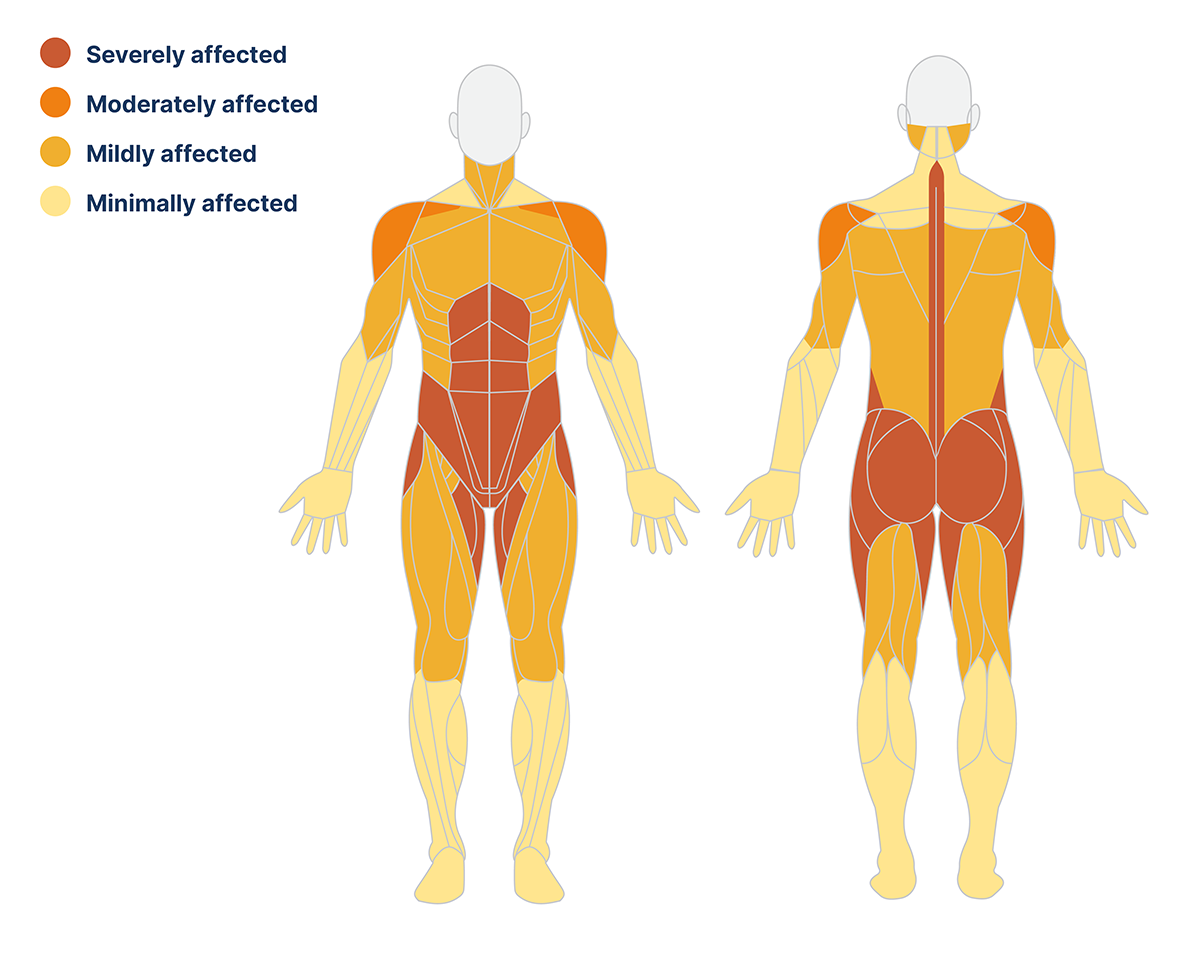
The pattern of muscle weakness is often symmetrical and does not differ between males and females. The limb-girdle and trunk muscles are affected early in the disease course, while the distal muscle groups are affected later or are not involved at all.9
Progressive muscle weakness in late-onset Pompe disease reduces the application of mechanical forces on the skeleton, which are essential for maintaining bone mineral density and strength.10 This in turn increases the risk of osteoporosis and bone fractures, especially when combined with physical impairment and a higher incidence of falls due to underlying myopathy.8,10
Glycogen also accumulates in smooth muscle cells and contributes to respiratory difficulties characteristic of late-onset Pompe disease.11 Please view the page on Respiratory manifestations above for further information.
If you suspect Pompe disease, referral to nominated ultra-specialist centre is essential so that patients can access full genetic testing and access to disease-specific therapies. Please refer to our ‘Find a specialist centre’ page to find out more.
NP-NN-GB-00031223
October 2024
- Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Curr Treat Options Neurol. 2022;24(11):573-588.
- Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics. 2018;15(4):928-942.
- Shah NM, Sharma L, Ganeshamoorthy S, Kaltsakas G. Respiratory failure and sleep-disordered breathing in late-onset Pompe disease: a narrative review. J Thorac Dis. 2020;12(Suppl 2):S235-S247.
- Taverna S, Cammarata G, Colomba P, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging. 2020;12(15):15856-15874.
- Lim JA, Li L, Kakhlon O, Myerowitz R, Raben N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy. 2015;11(2):385-402.
- Adeva-Andany MM, González-Lucán M, Donapetry-García C, Fernández-Fernández C, Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85-100.
- Katona I, Weis J, Hanisch F. Glycogenosome accumulation in the arrector pili muscle in Pompe disease. Orphanet J Rare Dis. 2014;9:17.
- Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-288.
- van der Beek NA, de Vries JM, Hagemans ML, et al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet Journal of Rare Diseases. 2012;7(1):88.
- Papadimas G, Terzis G, Papadopoulos C, et al. Bone density in patients with late onset Pompe disease. Int J Endocrinol Metab. 2012;10(4):599-603.
- Toscano A, Rodolico C, Musumeci O. Multisystem late onset Pompe disease (LOPD): an update on clinical aspects. Ann Transl Med. 2019;7(13):284.

0.
Neurology

2.
When should a neurologist suspect Pompe disease?