Respiratory
Respiratory pathophysiology in Pompe disease
The information on this page is tailored for respiratory specialists. For more detailed information on the cause, inheritance, diagnosis and management of Pompe disease, please refer to our ‘About Pompe disease’ page.
Pathology in late-onset Pompe disease arises from the abnormal accumulation of glycogen in the lysosomes of cells throughout various tissues in the body, particularly the skeletal muscle.1
This excessive glycogen storage triggers lysosomal rupture, autophagy and consequent muscle damage.1
Click on the headings to find out more.
Glycogen accumulation causes lysosomes to swell and rupture in the intermyofibrillar space of skeletal muscle. Glycogen and other enzymes leak into the cytoplasm of the myocytes, impairing their contractile function and leading to muscle damage and decline of muscle function.2,3
Normal lysosomal glycogen degradation begins with autophagy, a normal process by which the body consumes its own damaged tissue. Double-membrane vesicles called autophagosomes form and collect cellular contents, after which they fuse with lysosomes to begin the degradation process.4 This fusion is often defective in Pompe disease, resulting in the accumulation of autophagosomes and other debris that impact the contractility of affected skeletal muscle fibres.1,2,5
In late-onset Pompe disease, glycogen accumulation in the respiratory skeletal muscle impairs contractility and causes gradual muscle weakness. The diaphragm appears to be the most severely affected and its involvement is a hallmark of late-onset Pompe disease.3,6 The inspiratory and expiratory muscles – particularly those of the abdominal wall – are also impaired, along with the smooth muscle layers of the trachea and bronchi.7,8
Neural involvement in respiratory pathophysiology
Both human and animal models support the idea that neuropathology contributes to neuromuscular dysfunction and respiratory insufficiency in late-onset Pompe disease. This is strengthened by several patient case reports documenting glycogen accumulation in the central nervous system (CNS) and in phrenic motoneurons, key regulators of diaphragm expansion and contraction.6,9
This graph summarises one proposed model of the relationship between declines in muscular and neural function, and the associated impact on respiratory capabilities.6
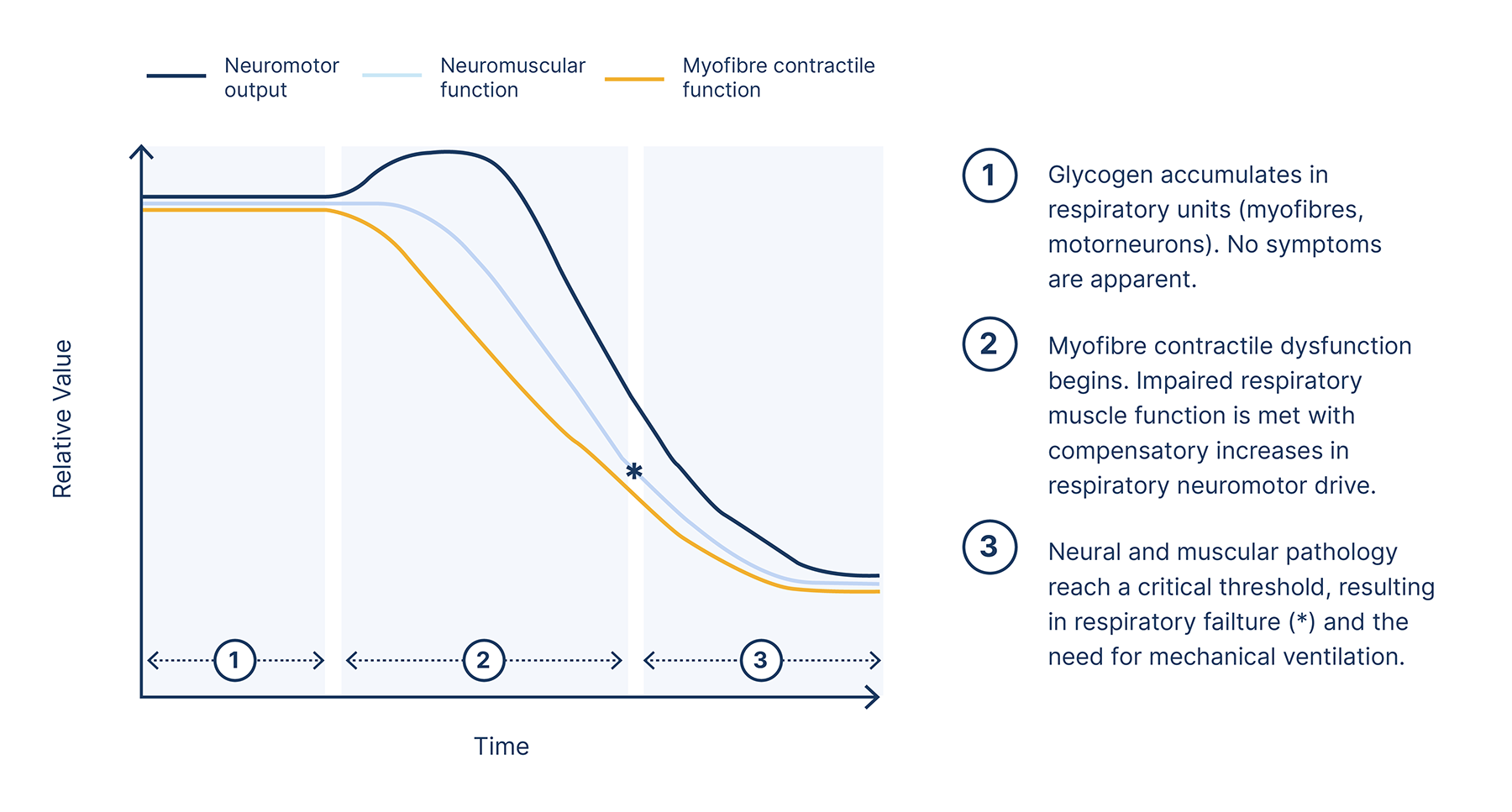
Reduced respiratory muscle capacity, reduced neural respiratory drive and increased load on the respiratory system will result in hypoventilation and eventual respiratory failure, with approximately 30–40% of late-onset Pompe disease patients requiring ventilation.3
Below, we see coronal slices through the carina for MRI breath-hold scans with the corresponding colour plots of a late-onset Pompe disease patient and a volunteer. Note the lack of diaphragm displacement in the Pompe disease patient. The colour maps represent the thickness of the segmentation in the anterior-posterior axis (red being the thickest and blue being the thinnest).10
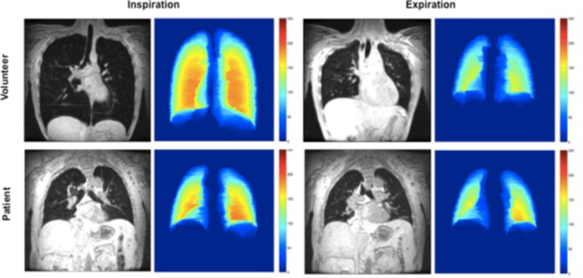
Wens SCA, et al. 2015, licensed under CC BY 4.0.10
If you suspect Pompe disease, referral to nominated ultra-specialist centre is essential so that patients can access full genetic testing and access to disease-specific therapies. Please refer to our ‘Find a specialist centre’ page to find out more.
NP-NN-GB-00021223
October 2024
- Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Curr Treat Options Neurol. 2022;24(11):573-588.
- Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics. 2018;15(4):928-942.
- Shah NM, Sharma L, Ganeshamoorthy S, Kaltsakas G. Respiratory failure and sleep-disordered breathing in late-onset Pompe disease: a narrative review. J Thorac Dis. 2020;12(Suppl 2):S235-S247.
- Taverna S, Cammarata G, Colomba P, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging. 2020;12(15):15856-15874.
- Lim JA, Li L, Kakhlon O, Myerowitz R, Raben N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy. 2015;11(2):385-402.
- Fuller DD, ElMallah MK, Smith BK, et al. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol. 2013;189(2):241-249.
- Spiesshoefer J, Henke C, Kabitz HJ, et al. The nature of respiratory muscle weakness in patients with late-onset Pompe disease. Neuromuscul Disord. 2019;29(8):618-627.
- Toscano A, Rodolico C, Musumeci O. Multisystem late onset Pompe disease (LOPD): an update on clinical aspects. Ann Transl Med. 2019;7(13):284.
- Falk DJ, Todd AG, Lee S, et al. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Human molecular genetics. 2015;24(3).
- Wens SC, Ciet P, Perez-Rovira A, Logie K, Salamon E, Wielopolski P, de Bruijne M, Kruijshaar ME, Tiddens HA, van Doorn PA, van der Ploeg AT. Lung MRI and impairment of diaphragmatic function in Pompe disease. BMC Pulm Med. 2015 May 6;15:54.

0.
Respiratory

2.
When should a respiratory specialist suspect Pompe disease?