About Pompe disease
GAA gene variants and inheritance
There are nearly 600 distinct variants in the GAA gene that have been linked to Pompe disease. Among these, some are considered pathogenic, while others are either benign or categorised as variants of unknown significance (VUS).1,2 All types of variants have been described with missense variants being the most frequent, followed by small deletions.3
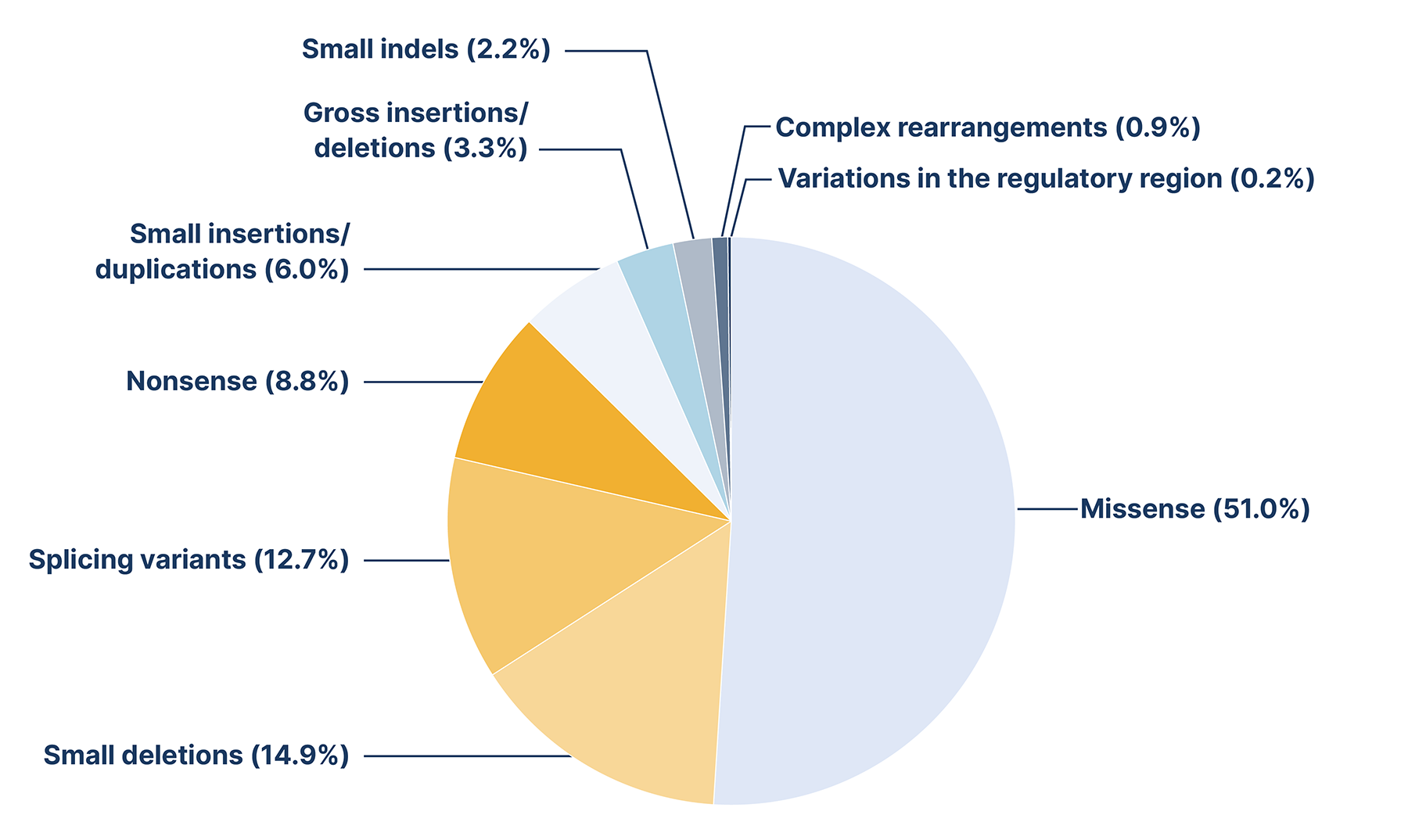
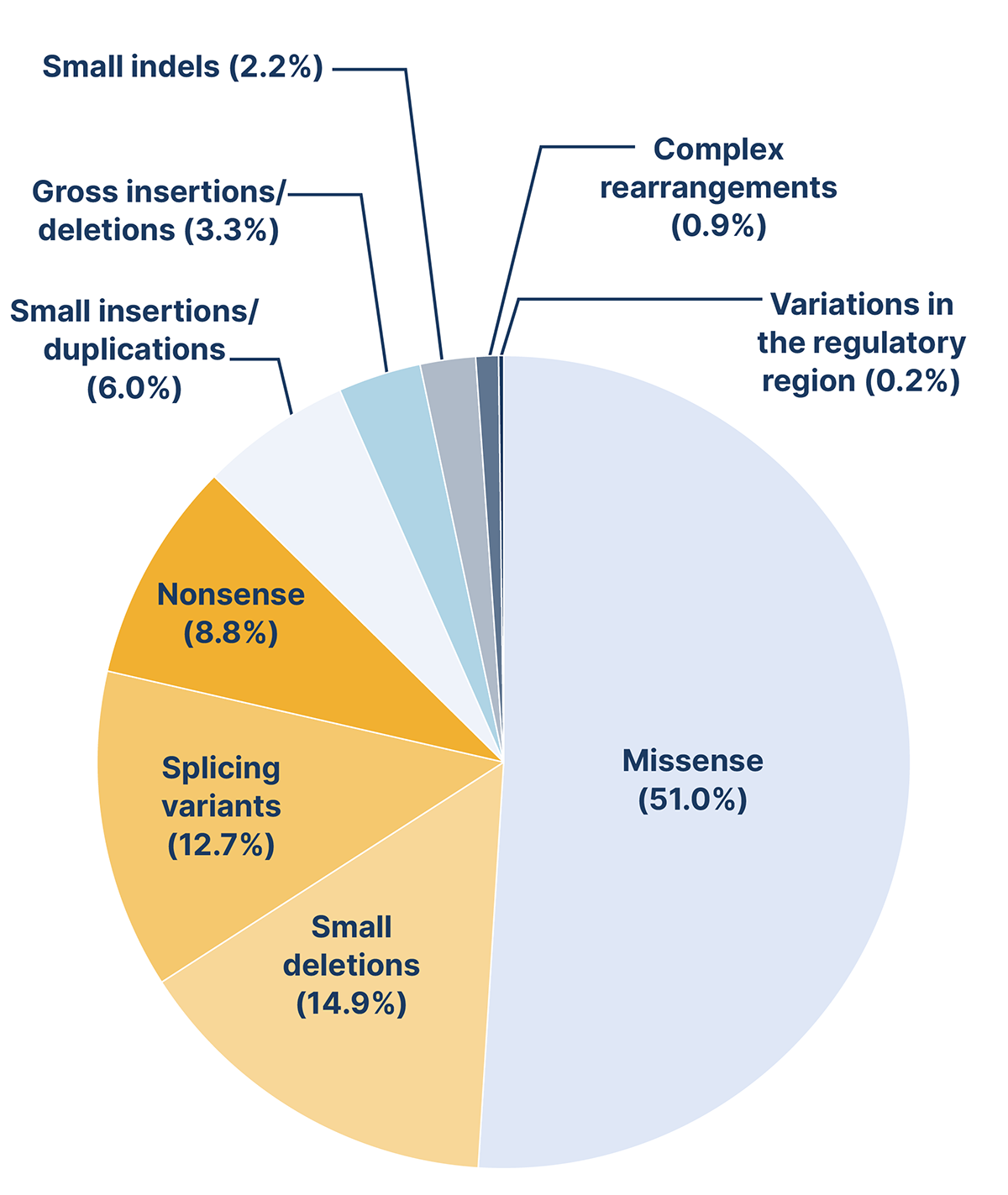
Frequency of 582 GAA variants reported in the Human Gene Mutation Database (HMGD®) in 2019. Adapted from Peruzzo et al., 2019.3
Genotype-phenotype correlation
In general, patients affected by infantile-onset Pompe disease possess pathogenic GAA variants that severely reduce the expression of the functional GAA protein. The presence of a potentially mild variant in one allele seems to prevent the occurrence of a severe infantile-onset phenotype.3 However, as with most genetic diseases, a strict correlation between the phenotype and genotype of Pompe disease is difficult to establish, primarily because most GAA variants are unique and are specific to a small number of families.3
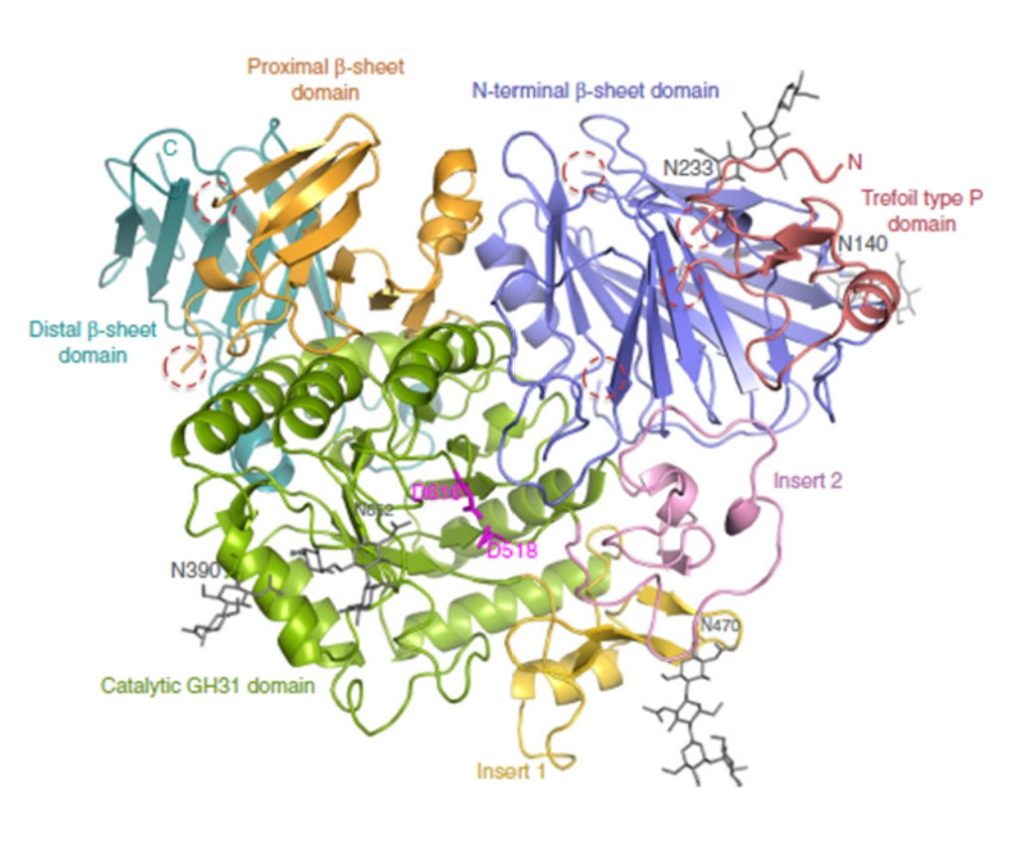
Roig-Zamboni et al., 2017, licensed under CC BY 4.0.4
The one exception to this rule is the c.-32-13T>G splice variant, which is very common in patients of Caucasian origin affected by the late-onset form of disease. Its frequency ranges from 40–70% in different populations.3 Studies display a wide variability in residual GAA activity, age at onset and disease progression associated with this variant. Phenotypic differences have also been reported in siblings carrying this genotype.3
From these findings, it can be concluded that secondary factors, both genetic and non-genetic, play a role in modifying Pompe disease phenotype.3
Inheriting Pompe disease
Pompe disease is inherited in an autosomal recessive pattern. This means individuals must receive two GAA variants, one from each parent, to inherit the condition. The risk is equal for both males and females.1
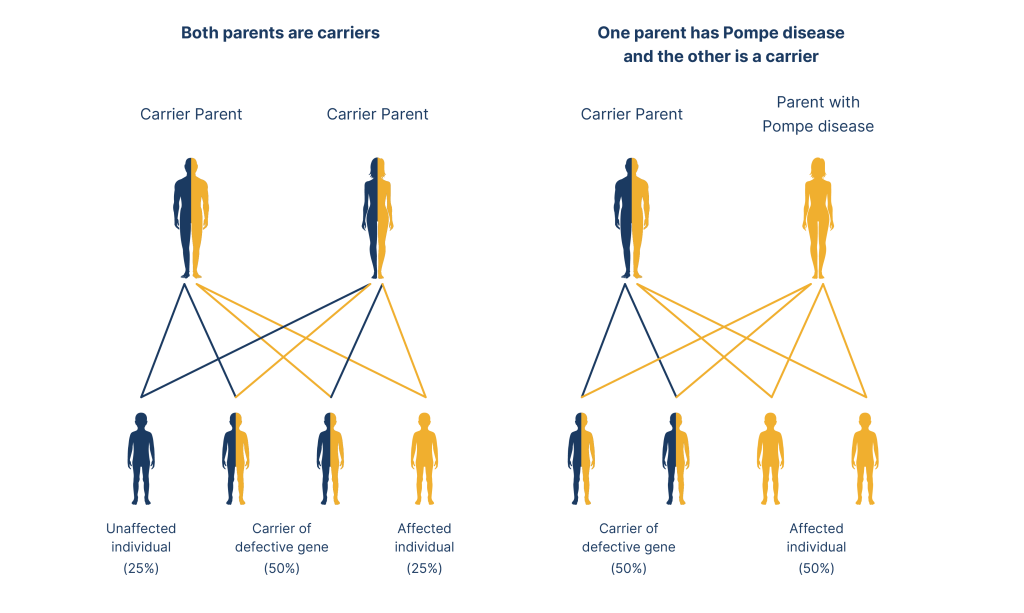
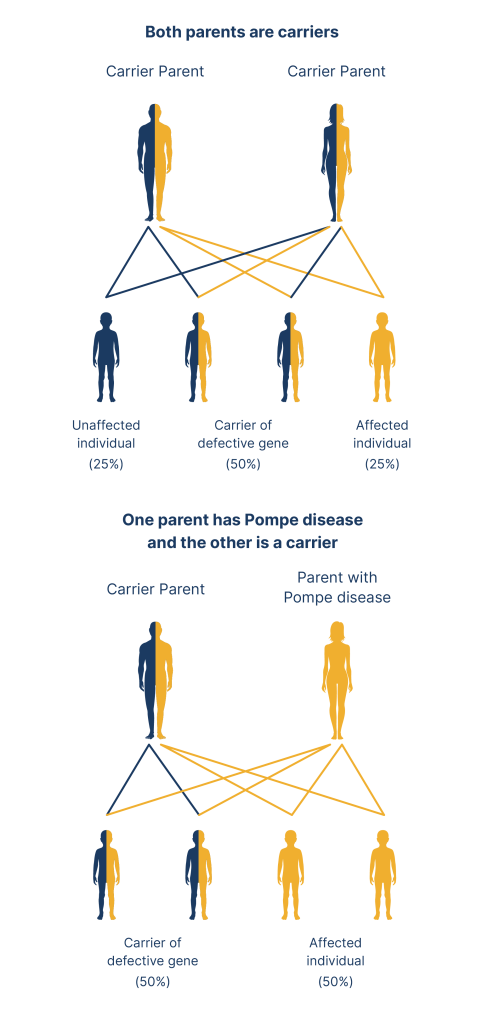
Adapted from Wexler, M. 2023.5
Genetic counselling
Genetic counselling is offered to individuals and families with a Pompe disease diagnosis to help them understand the nature, inheritance and implications of genetic disorders and inform their medical and personal decisions. Carrier testing of other family members using a biochemical or genetic approach is also appropriate, alongside prenatal testing for pregnancies at increased risk (in families where the disease-causing variant is already known).6
Certain requesting specialties can arrange genetic testing to confirm a suspected Pompe disease diagnosis. Individuals diagnosed with Pompe disease should then be referred to a nominated ultra-specialist centre to access disease-specific therapies. Please refer to our ‘Genetic testing for Pompe disease’ page to find out more.
Com-NN-UKI-26-00019
May 2026
- NORD. Pompe Disease. 2024. Available at: https://rarediseases.org/rare-diseases/pompe-disease/. Accessed: May 2026.
- Reuser AJJ, van der Ploeg AT, Chien YH, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum Mutat. 2019;40(11):2146-2164.
- Peruzzo P, Pavan E, Dardis A. Molecular genetics of Pompe disease: a comprehensive overview. Ann Transl Med. 2019;7(13):278.
- Roig-Zamboni V, Cobucci-Ponzano B, Iacono R, et al. Structure of human lysosomal acid α-glucosidase-a guide for the treatment of Pompe disease. Nat Commun. 2017;8(1):1111.
- Wexler M. How is Pompe disease inherited? April 2023. Available at: https://pompediseasenews.com/pompe-inheritance/. Accessed: May 2026.
- Taglia A, Picillo E, D’Ambrosio P, Rosaria Cecio M, Viggiano E, Politano L. Genetic counseling in Pompe disease. Acta Myol. 2011;30(3):179-181.

2.
Prevalence of Pompe disease

4.
Clinical manifestations of Pompe disease